Polymorphism detection and genotyping¶
Introduction¶
Adapative immune receptor repertoire sequencing (AIRR-Seq, Rep-Seq) data is currently the subject of much study. A key step in analyzing these data involves assigning the closest known V(D)J germline alleles to the (often somatically mutated) sample sequences using a tool such as IMGT/HighV-QUEST (1). However, if the sample utilizes alleles not in the germline database used for alignment, this step will fail. Additionally, this alignment has an associated error rate of ~5% (2), notably among sequences carrying a large number of somatic mutations.
Here we provide a Tool for Immunoglobulin Genotype Elucidation via Rep-Seq (TIgGER). TIgGER addresses these issues by inferring the set of Immunoglobulin (Ig) alleles carried by an individual (including any novel alleles) and then using this set of alleles to correct the initial assignments given to sample sequences by existing tools.
This vignette covers the following tasks:
- Inferring the presence of novel IGHV alleles not in the germline database.
- Inferring the personalized IGHV genotype of a sample.
- Correcting the IGHV allele calls of a sample based on the IGHV genotype.
Additional information about the methods used by TIgGER is available in:
Input¶
TIgGER requires two main inputs:
- Pre-processed Ig sequence data
- Database germline sequences
AIRR-seq data is input as a data frame following the AIRR or Change-O standard where each row represents a unique observation and and columns represent data about that observation. The required names of the required columns are provided below along with a description of each.
| Column Name | Description |
|---|---|
SEQUENCE_IMGT or sequence_alignment |
V(D)J sequence gapped in the IMGT gapped format (3) |
V_CALL or v_call |
(Comma separated) name(s) of the nearest V allele(s) |
J_CALL or j_call |
(Comma separated) name(s) of the nearest J allele(s) |
JUNCTION or junction |
Junction nucleotide sequence |
JUNCTION_LENGTH or junction_length |
Length of the junction region of the V(D)J sample |
An example dataset is provided with the tigger package as SampleDb (Change-O format)
and AIRRDb (AIRR format). It contains unique functional sequences assigned to IGHV1 family genes
isolated from individual PGP1 (referenced in Gadala-Maria et al. 2015).
The database of germline sequences should be provided in FASTA format with
sequences gapped according to the IMGT numbering scheme (3). IGHV alleles in
the IMGT database (build 2014-08-4) are provided with this package as SampleGermlineIGHV.
You may read in your own fasta file using readIgFasta.
# Load packages required for this example
library(tigger)
library(dplyr)
Novel allele detection¶
Potential novel alleles can be detected by TIgGER. Some of these may be included
in the genotype later (see below). findNovelAlleles will return a data.frame
with a row for each allele tested for the presence of polymorphisms. If
polymorphisms are found and the novel sequence passes all quality tests (see
below), then the novel allele is named and the germline sequence is included in
the data.frame. Additionally, the result will contain metadata on the
parameters used when searching for novel alleles (which can be optionally
changed in findNovelAlleles).
# Detect novel alleles
novel <- findNovelAlleles(AIRRDb, SampleGermlineIGHV, nproc=1)
# Extract and view the rows that contain successful novel allele calls
novel_rows <- selectNovel(novel)
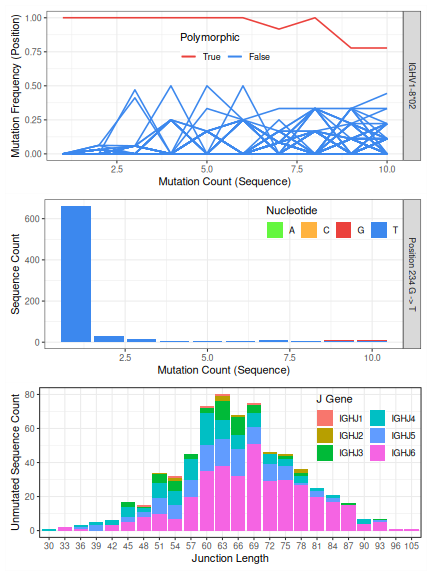
The TIgGER procedure for identifying novel alleles (see citation above) involves
taking all sequences which align to a particular germline allele and, for each
position along the aligned sequences, plotting the mutation frequency at that
postion as a function of the sequence-wide mutation count. While mutational
hot-spots and cold-spots are both expected to have a y-intercept around zero,
polymorphic positions will have a y-intercept larger than zero. The required
minimum y-intercept may be specified in findNovelAlleles by y_intercept, but
defaults to 1/8. Passing this y-intercept threshold is the first of three
pieces of evidence that support the novel allele.
The second piece of evidence supporting novel allele calls is the nucleotide usage at the polymorphic positions as a function of sequence-wide mutation count. We expect the polymorphic allele to be prevalent at all mutation counts, and we expect the mutation count equal to the number of polymorphisms in the novel sequence to be the most prevalent.
Finally, to avoid cases where a clonal expansion might lead to a false positive,
combinations of J gene and junction length are examined among sequences which
perfectly match the proposed germline allele. A true novel allele is expected
to utilize a wide range of J genes, and sequences with different junction length
can be ruled out as not being clonally related. The maximum portion of sequences
which can consist of a specific combination of J gene and junction length may be
specified in findNovelAlleles by j_max.
The three pieces of evidence desribed above can be viewed for any allele call
made by findNovelAlleles using the function plotNovel.
# Plot evidence of the first (and only) novel allele from the example data
novel_row <- which(!is.na(novel$polymorphism_call))[1]
plotNovel(AIRRDb, novel[novel_row, ])
Inferring genotypes¶
An individual’s genotype can be inferred using the functions inferGenotype or
inferGenotypeBayesian. Using one of this functions allows to remove from the
genotype rare/erroneous allele calls which may result from mutations in
allele-differentiating regions. inferGenotype uses a frequency method to
decide which alleles belong to the subjects genotype whereas
inferGenotypeBayesian infers an subject’s genotype applying a Bayesian
framework and provides a confidence estimate associated with
the genotype calls.
Frequency genotyping approach¶
inferGenotype identifies the fewest alleles that account for
nearly all (default is 7/8) of the allele calls made. The user may opt to only
use sequences which perfectly match germline alleles, and may opt to include
potential novel alleles. (The genotype output is designed to be human readable,
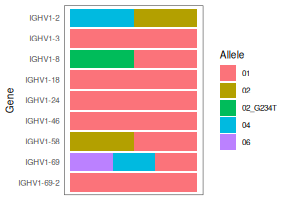
though plotGenotype can be used to make a colorful visualization.) For each
allele, the number of sequences which match the germline are listed in the same
order as the alleles are listed. The total number of sequences that match any
allele of that gene is also given. To output these alleles as a names vector of
nucleotide sequences, the user may use the function genotypeFasta. To save
this vector to a fasta file, writeFasta may be used.
# Infer the individual's genotype, using only unmutated sequences and checking
# for the use of the novel alleles inferred in the earlier step.
geno <- inferGenotype(AIRRDb, germline_db=SampleGermlineIGHV, novel=novel,
find_unmutated=TRUE)
# Save the genotype sequences to a vector
genotype_db <- genotypeFasta(geno, SampleGermlineIGHV, novel)
# Visualize the genotype and sequence counts
print(geno)
## gene alleles counts total
## 1 IGHV1-2 02,04 664,302 966
## 2 IGHV1-3 01 226 226
## 3 IGHV1-8 01,02_G234T 467,370 837
## 4 IGHV1-18 01 1005 1005
## 5 IGHV1-24 01 105 105
## 6 IGHV1-46 01 624 624
## 7 IGHV1-58 01,02 23,18 41
## 8 IGHV1-69 01,04,06 515,469,280 1279
## 9 IGHV1-69-2 01 31 31
## note
## 1
## 2
## 3
## 4
## 5
## 6
## 7
## 8 Cannot distinguish IGHV1-69*01 and IGHV1-69D*01
## 9
# Make a colorful visualization. Bars indicate presence, not proportion.
plotGenotype(geno, text_size = 10)
Bayesian genotyping approach¶
The method inferGenotypeBayesian analyzes the posterior probabilities of
possible allele distributions, considering up to four distinct alleles per
V gene, corresponding to a gene duplication with both loci being heterozygous
(i.e., homozygous, heterozygous with one copy of each allele, etc.). The
posterior probabilities for these four possible models are compared and a Bayes
factor is calculated for the two most probable models. This Bayes factor
reflects the confidence in the genotyping call of the method. The bayesian
method doesn’t use the strict cutoff criterion fraction_to_explain that
inferGenotype uses wherein only the minimum set of alleles explaining
88% (7/8) of apparently-unmutated sequences are included in the genotype.
# Infer the individual's genotype, using the bayesian method
geno_bayesian <- inferGenotypeBayesian(AIRRDb, germline_db=SampleGermlineIGHV,
novel=novel, find_unmutated=TRUE)
# Visualize the genotype and sequence counts
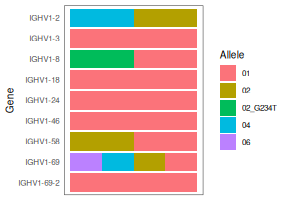
print(geno_bayesian)
## gene alleles counts total
## 1 IGHV1-2 02,04 664,302 966
## 2 IGHV1-3 01 226 226
## 3 IGHV1-8 01,02_G234T 467,370 837
## 4 IGHV1-18 01 1005 1005
## 5 IGHV1-24 01 105 105
## 6 IGHV1-46 01 624 624
## 7 IGHV1-58 01,02 23,18 41
## 8 IGHV1-69 01,04,06,02 515,469,280,15 1279
## 9 IGHV1-69-2 01 31 31
## note kh
## 1 -1000
## 2 4.20089197988625
## 3 -1000
## 4 -3.76643736033536
## 5 4.75335701924247
## 6 0.457455409315221
## 7 -20.3932114156223
## 8 Cannot distinguish IGHV1-69*01 and IGHV1-69D*01 -1000
## 9 4.16107190423977
## kd kt kq k_diff
## 1 -7.92846809405969 -139.556367176944 -313.583949130729 131.627899082884
## 2 -45.2911957825576 -84.2865868763307 -128.991761853586 49.4920877624439
## 3 -1.04759115960507 -102.524664723923 -247.193958844361 101.477073564318
## 4 -223.85293382607 -1000 -1000 220.086496465735
## 5 -18.2407545518045 -36.3580822723628 -57.1281856909991 22.9941115710469
## 6 -136.193264784335 -243.861955237939 -1000 136.65072019365
## 7 3.60009261357983 -1.38512929425796 -8.47869574581951 4.9852219078378
## 8 -277.291087469703 3.55051520054669 -143.380669247128 146.931184447674
## 9 -2.62766579768837 -7.97659112471034 -14.1087168959268 6.78873770192814
# Make a colorful visualization. Bars indicate presence, not proportion.
plotGenotype(geno_bayesian, text_size=10)
Correcting allele calls¶
Finally, the original V allele calls may be limited to only those within the
inferred genotype. This can be done by using the function reassignAlleles.
By correcting the calls in this manner, the user can greatly reduce the numer of
ambiguous allele calls (where a single sample sequences is assigned to multiple
V alleles, thus preventing the mutations analysis of allele-differentiating
positions). Additionally, assignments to erroneous not-in-genotype alleles
(expected to be ~5% (2), as mentioned above, are corrected in this manner.
# Use the personlized genotype to determine corrected allele assignments
# Updated genotype will be placed in the v_call_genotyped column
sample_db <- reassignAlleles(AIRRDb, genotype_db)
From here, one may proceed with further downstream analyses, but with the advantage of having much-improved allele calls. Besides having discovered alleles not in the IMGT database, the calls made by IMGT have been tailored to the subject’s genotype, greatly reducing the number of problematic calls, as can be seen below.
# Find the set of alleles in the original calls that were not in the genotype
not_in_genotype <- sample_db$v_call %>%
strsplit(",") %>%
unlist() %>%
unique() %>%
setdiff(names(genotype_db))
# Determine the fraction of calls that were ambigious before/after correction
# and the fraction that contained original calls to non-genotype alleles. Note
# that by design, only genotype alleles are allowed in "after" calls.
data.frame(Ambiguous=c(mean(grepl(",", sample_db$v_call)),
mean(grepl(",", sample_db$v_call_genotyped))),
NotInGenotype=c(mean(sample_db$v_call %in% not_in_genotype),
mean(sample_db$v_call_genotyped %in% not_in_genotype)),
row.names=c("Before", "After")) %>%
t() %>% round(3)
## Before After
## Ambiguous 0.112 0.15
## NotInGenotype 0.057 0.00
Evidence¶
generateEvidence uses the final corrected calls, the novel alleles and genotype
information, the final genotype sequences and the starting reference germlines
to build a table of evidence metrics supporting the final novel V allele detection.
evidence <- generateEvidence(sample_db, novel, geno, genotype_db, SampleGermlineIGHV, fields = NULL)
evidence %>%
select(gene, allele, polymorphism_call, sequences, unmutated_frequency)
## # A tibble: 1 × 5
## # Rowwise:
## gene allele polymorphism_call sequences unmutated_frequency
## <chr> <chr> <chr> <int> <dbl>
## 1 IGHV1-8 02_G234T IGHV1-8*02_G234T 864 0.428
In this example, 864 sequences were unambiguously assigned to allele IGHV1-8*02_G234T, 42.82% of them unmutated.